Sindrome di Behçet: quali sono i sintomi e la terapia
La sindrome di Behcet è una vasculite sistemica, multisintomatica con decorso cronico. Essa è stata descritta per la prima volta nel 1937 dal dermatologo Behcet. Si caratterizza per la presenza di una triade:
- Ulcereazioni orali (afte orali);
- Ulcere genitali (afte genitali);
- Ulcere oculari (1).
Indice delle informazioni che troverai nell’articolo
Sindrome di Behcet: quali distretti colpisce
La malattia di behcet presenta queste tre ulcere in concomitanza, infatti, la triade viene definita contemporanea.

By Adert – Own work, CC BY-SA 3.0, https://commons.wikimedia.org/w/index.php?curid=32460792
Oltre a queste tre manifestazioni, la malattia può colpire la cute nel 90% dei bambini colpiti (eritema nodoso in sede pretibiale), il sistema nervoso attraverso l’encefalomielite, il sistema vascolare colpendo sia vene che arterie di qualsiasi calibro e il sistema gastrointestinale con dolori addominali, perdita di peso, diarrea, le grandi articolazioni come il ginocchio, la caviglia, il gomito e il polso. Dolori articolari che non si presentano in maniera bilaterale. Molto più raramente colpisce anche il sistema cardiovascolare e renale (2).
Afte orali
Le ulcere orali, in genere, sono i primi segni della malattia. Si possono presentare in svariati modi e luoghi. Possono essere superficiali o profonde e possono interessare la lingua, la mucosa orale o la gola. Generalmente le afte orali si ripresentano ciclicamente senza lasciare particolari cicatrici (2).
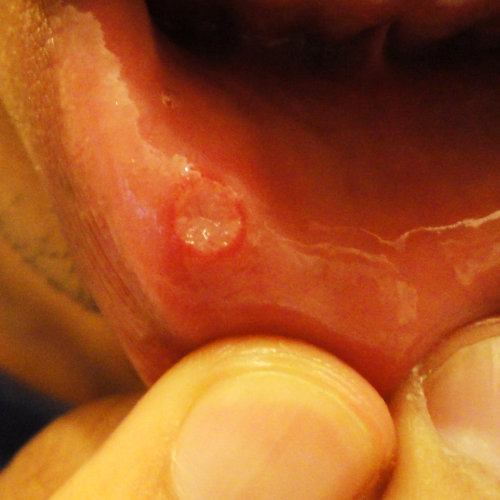
In questa foto possiamo visualizzare un afta orale presente sul labbro inferiore
Ulcere genitali
A differenza delle afte del cavo orale, le ulcere genitali, spesso, lasciano cicatrici e si presentano nell’uomo maggiormente sullo scroto (sacca che contiene i testicoli), nella donna maggiormente sulle grandi labbra (2).
Ulcere oculari
Le ulcere possono coinvolgere sia la retina che l’uvea. L’uveite è una manifestazione molto diffusa. Il quadro clinico portato dalle ulcere oculari è molto grave poiché le cicatrici possono causare l’annebbiamento o perdita della vista. Esse sono più diffuse nei bambini e colpiscono maggiormente il sesso maschile (2).

Di EyeMD (Rakesh Ahuja, M.D.). – Opera propria., CC BY-SA 2.5, https://commons.wikimedia.org/w/index.php?curid=1229578
Malattia di Behcet: incidenza
In alcune zone, per esempio in Turchia, il morbo di Behcet presenta delle incidenze altissime. Secondo i dati riportati su Orphanet, la sindrome colpisce circa 1 persona su 1.000 (3). Il range di età è ampio ed è compreso tra i 5 e i 55 anni.
Morbo di Behcet: cause
La causa della sindrome di Bechet è ancora sconosciuta. Si pensa che l’origine sia di carattere autoimmune e che comprenda un insieme di fattori scatenanti come la genetica, infettiva o immunitaria. Si ha una familiarità importante nel contrarre la malattia. Alcuni studi hanno riportato la possibilità che un agente infettivo possa scatenare la malattia, ma la presa in considerazione dell’herpes simplex, spesso presente in concomitanza del morbo di bechet, o di batteri come lo Streptococco, non hanno riportato un diretto legame con la patologia. Detto questo, le poche informazioni portano ad indicare l’origine della malattia come multifattoriale (2).
Sintomi
I sintomi della malattia di Behcet sono rappresentati principalmente da infiammazione diffusa all’encefalo con interessamento delle meningi (meningoencefalite), che si manifesta in:
- Cefalea;
- Confusione;
- Febbre.
Solo nel 15% dei casi, la malattia di Bechet colpisce il sistema nervoso centrale, causando non pochi e leggeri sintomi. Infatti, in rari casi può colpire il tronco cerebrale andando a ad interessare l’emergenza dei nervi cranici con tutte le conseguenze del caso (2-4).
Diagnosi
La diagnosi del morbo di behcet ha un carattere puramente clinico poiché non esistono esami ematochimici specifici che possano dare indicazione sulla presenza della malattia. Sicuramente, nella forma acuta, si assiste ad un aumento della presenza dei fattori dell’infiammazione con un aumento delle citochine circolanti che hanno un valore aspecifico a differenza della IL-17, citochina presente in presenza della malattia. Si associa, durante la malattia, anche elevati valori del fattore di von Willebrand. Nonostante la presenza di tali valori, la diagnosi della malattia di Behcet arriva dalla presenza di afte orali ricorrenti, almeno 3 nell’arco di 12 mesi. Le afte devono necessariamente essere accompagnate dalla presenza di almeno due dei seguenti segni: ulcere cutenee, ulcere oculari, ulcere genitali (2-3).
Terapia
La terapia della sindrome di behcet varia in base alla localizzazione della malattia. Tutti i pazienti comunque presentano una terapia steroidea antinfiammatoria. Come già specificato in base alla sede colpita, la terapia viene integrata con l’assunzione di altri farmaci. Per esempio, l’assunzione del prednisone viene eseguita in situazioni in cui la sindrome colpisce il sistema nervoso centrale, l’assunzione di eparina viene eseguita nei casi in cui il morbo colpisce il sistema vascolare (trombosi). L’uso di farmaci immunosoppressori, vengono usati quando le manifestazioni della malattia sono sistemiche (1).
- Angelo Sghirlanzoni, Terapie Delle Malattie Neurologiche. Springer Verlag, ed. 2010;
- Vincenzo Zuccotti, Manuale di pediatria. La pratica clinica. Esculapio; 2° edizione 2016;
- Orphanet;
- Monti, L’ulcera cutanea. Approccio multidisciplinare alla diagnosi e al trattamento: Approccio multidisciplinare alla diagnosi ed al trattamento. Springer Verlag, 1° ed. 2000.





0 commenti